Neurologia e Doenças Raras - Doença de Fabry
O que é a doença de Fabry?
Também conhecida como doença de Anderson-Fabry, foi descrita pela primeira vez em 1898. É um distúrbio genético progressivo associado ao cromossomo X, podendo afetar tanto pessoas do sexo masculino quanto feminino. Ela é considerada uma das doenças raras em neurologia, com incidência de cerca de 1:40.000 nos homens e 1:20.000 nas mulheres. (Clique aqui e saiba o conceito de doença rara).
Evolução
A doença pode comprometer o paciente de forma grave e seus sintomas não são específicos, podendo dificultar o diagnóstico.
Causas
Distúrbio de armazenamento lisossômico vinculado ao X. Ocorre mutação no gene GLA no cromossomo X, causando deficiência da alfa-galactosidase A (alga-Gal A), resultando no acúmulo progressivo de globotriaosilceramida nos lisossomos, o que causa dano a diversos órgãos. (1)
É comum que exista relato de história familiar de problemas renais sem causa identificada ou relato de Ataque isquêmico transitório (AIT), Acidente Vascular Cerebral isquêmico (AVCi) ou Infarto Agudo do Miocárdio (IAM) em familiares jovens (antes dos 55 anos).
Sinais e Sintomas
Pode afetar diversos órgãos e sistemas:
Cardio: anormalidades de condução, arritmia, hipertrofia do ventrículo esquerdo, coronariopatia precoce, angina, infarto do miocárdio, prolapso de válvula mitral e/ou regurgitação.
Rins: cistos, albuminúria, proteinúria, insuficiência renal progressiva de etiologia desconhecida; disfunção tubular (poliúria, polidipsia), sintomas que sugerem síndrome de Fanconi, aumento de creatinina sérica.
Gastrointestinal: dor abdominal, náuseas, vômitos, constipação, diarreia.
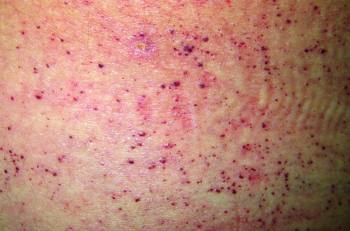
Pele: angioqueratomas, telangiectasias.
Olhos: córnea verticilata, tortuosidades dos vasos retinianos.
Orelhas: comprometimento auditivo, vertigem, tinido.
Sistema nervoso: AIT, AVCi, distúrbios neuropsiquiátricos, acroparestesia (principalmente em mãos e pés), hipohidrose, hipersensibilidade às mudanças térmicas (calor ou frio).
Reumato: dores articulares, intolerância a exercícios físicos, angioqueratomas, febre, VHS elevado.
Apresentação
Na forma clássica da doença de Fabry os sintomas normalmente aparecem na infância com dor neuropática crônica, crises de dor aguda do tipo acroparestesia, intolerância ao calor ou ao frio e fadiga. (2)
Com a progressão do quadro o paciente passa a apresentar outros sintomas nos sistemas descritos acima na seção de sinais e sintomas.
Diagnóstico
O diagnóstico costuma ser dificultado, pois a evolução clínica é variável e a doença pode afetar diversos sistemas.
O tempo médio entre o início dos sintomas e o diagnóstico é de 13,7 anos para os homens adultos e 16,3 anos para as mulheres adultos (3). Esse grande intervalo de tempo acarreta um prognóstico reservado entre os pacientes. O diagnóstico precoce é fundamental para o controle da doença.
Nos homens o diagnóstico pode ser feito pelos níveis da atividade da enzima alfa-Gal A nos leucócitos ou em amostras de sangue seco. O diagnóstico confirmatório é feito pelo sequenciamento do gene GLA.
ATENÇÃO: No caso de mulheres afetadas (heterozigotas para o gene mutado) o ensaio de atividade enzimática não é confiável. Neste caso é necessário o teste genético do GLA.
Tratamento
Infelizmente, até o momento, a doença de Fabry não é curável, mas pode ter algum controle com o tratamento específico que são as terapias de reposição enzimática (TRE) associadas ao tratamento de controle dos sintomas.
A TRE serve como uma fonte exógena da enzima deficitária e ajuda a retardar a progressão da doença.
Atualmente existem duas opções de TRE para os pacientes com diagnóstico conformado: via intravenosa em semanas alternadas.
Aconselhamento
Em caso de diagnóstico de doença de Fabry é recomendado que o paciente seja encaminhado para o geneticista para aconselhamento genético.
Referências:
Schäfer E, Baron K, Widmer U, Deegan P, Neumann HP, Sunder-Plassmann G, Johansson JO, Whybra C, Ries M, Pastores GM, Mehta A, Beck M, Gal A. Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease. Hum Mutat. 2005 Apr;25(4):412. doi: 10.1002/humu.9327. PMID: 15776423.
Mehta A, Beck M, Eyskens F, Feliciani C, Kantola I, Ramaswami U, Rolfs A, Rivera A, Waldek S, Germain DP. Fabry disease: a review of current management strategies. QJM. 2010 Sep;103(9):641-59. doi: 10.1093/qjmed/hcq117. Epub 2010 Jul 21. PMID: 20660166.
Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, Linhart A, Sunder-Plassmann G, Ries M, Beck M. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004 Mar;34(3):236-42. doi: 10.1111/j.1365-2362.2004.01309.x. PMID: 15025684.
Texto escrito por Dra. Letícia Januzi CRM AL 5647/ RQE 3137
Data: 05-02-2023
Última modificação: 05-02-2023

Sobre
Atua como médica neurologista especialista em AVC ("derrame cerebral")
Professora da FAMED-UFAL
CRM - AL 5647
RQE 3137



